



業(yè)務(wù)咨詢
中國:
Email: marketing@medicilon.com.cn
業(yè)務(wù)咨詢專線:400-780-8018
(僅限服務(wù)咨詢,其他事宜請撥打川沙總部電話)
川沙總部電話: +86 (21) 5859-1500
海外:
+1(781)535-1428(U.S.)
0044 7790 816 954 (Europe)
Email:marketing@medicilon.com




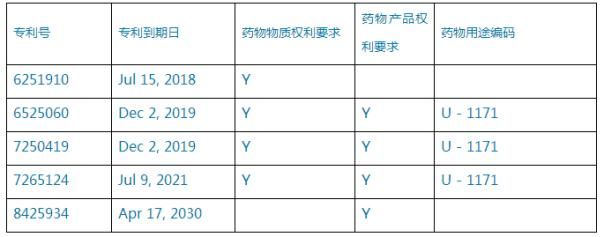
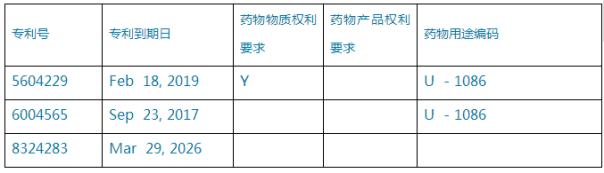
 相關(guān)新聞
相關(guān)新聞川沙總部
地址: 上海市浦東新區(qū)川大路585號
郵編: 201299
電話: +86 (21) 5859-1500(總機)
傳真: +86 (21) 5859-6369
業(yè)務(wù)咨詢
中國:
Email: marketing@medicilon.com
業(yè)務(wù)咨詢專線:400-780-8018
(僅限服務(wù)咨詢,其他事宜請撥打川沙
總部電話)
海外:
Email:?marketing@medicilon.com
Tel: +1 (617) 888-9294(U.S.)
Tel: 0044 7790 816 954 (Europe)
Tel: +82 70-8269-5849 (Korea)
Tel: +81 80-4421-6898 (Japan)











